New stem cell research reveals dangerous cardiac effects of a gene mutation in patients with Dravet syndrome.
1:00 PM
Author |

Imagine putting your child to bed, only to have him die inexplicably in his sleep.
This is the chilling reality for many victims of sudden unexpected death in epilepsy, or SUDEP — which claims the lives of roughly 1 in every 1,000 people with epilepsy or other seizure disorders.
LISTEN UP: Add the new Michigan Medicine News Break to your Alexa-enabled device, or subscribe to our daily audio updates on iTunes, Google Play and Stitcher.
Patients with a rare disease called Dravet syndrome are at heightened risk of SUDEP. In the disease, seemingly healthy infants develop frequent and prolonged seizures, catastrophically affecting their development and quality of life.
Michigan Medicine scientists have been on a quest to better understand the connection and potential ways to prevent SUDEP in this population.
What they know: Eighty percent of patients with Dravet syndrome have variants in the SCN1A gene, which codes for Nav1.1 sodium channels in the heart and brain. A sodium channel is like a gate that allows sodium ions across a cell membrane, producing an electrical charge.
SCN1A mutations in people with Dravet syndrome result in electrical disturbances inside cells. Although seizures are the result of abnormal electrical signals in the brain, researchers suspected that problems may also arise in the heart.
"We had a hypothesis that since these kids have the same mutation in their sodium channels in the heart and brain, they might have cardiac arrhythmias," says Lori Isom, Ph.D., chair of the Department of Pharmacology at Michigan Medicine. "We were able to gather evidence that they do."
The work is published in Stem Cell Reports.
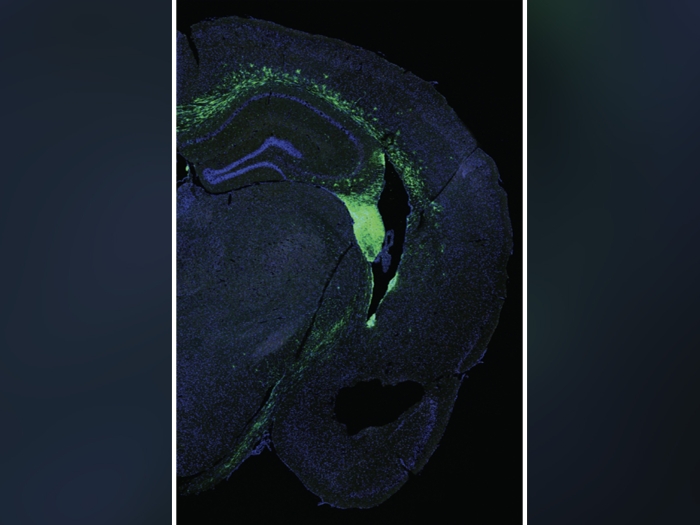
Isom and clinical colleague Jack M. Parent, M.D., professor of neurology and co-director of the Comprehensive Epilepsy Program at the University of Michigan, first looked to mouse models and then at cells collected from children with Dravet syndrome.
They discovered that mutations associated with Dravet syndrome in mice led to irregularities in the heart muscle's sodium channels.
And those irregularities could be fatal. Variants in sodium channels in the heart can cause ventricular arrhythmias, which occur when the lower chambers of the heart begin to beat abnormally.
What stem cells can tell
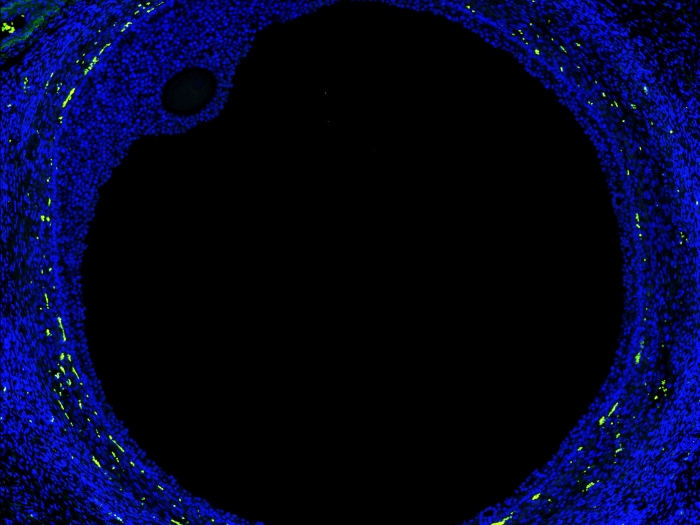
To see if that finding appeared in people, Isom and Parent collected skin cells from pediatric patients with Dravet syndrome all over the world. They converted those cells into induced pluripotent stem cells, which have the capacity to become any cell in the body.
MORE FROM THE LAB: Subscribe to our weekly newsletter
By turning them into cardiac cells, the researchers were able to show that despite the loss of the SCN1A gene, there was an increase in sodium current in the heart cells.
"Your body needs to maintain homeostasis. … It doesn't just stand there and take the insult, it does something in response," Isom explains. "So what the cell does to try and right the ship, so to speak, is to increase the expression of another sodium channel that's not mutated. But that appears to result in an uncontrolled overexpression, which produces too much sodium current."
In fact, Isom recalls that she and Parent sounded an alarm when looking at the cells of one patient: "When we saw a huge increase in sodium current, we looked at each other and said: 'If this was our child, we'd want to know about it.'"
They recommended that the child get a full cardiac workup to check for abnormalities, which turned out to be present.
[W]e looked at each other and said: 'If this was our child, we'd want to know about it.'Lori Isom, Ph.D.
'Personalized' approach serves many
Although heart arrhythmias may help explain the suddenness of SUDEP, uncertainties remain.
"The question is, why do you die after the 1,000th seizure when you didn't die in the 999th?" Isom says. "Sodium channel-linked cardiac arrhythmias almost always happen in sleep. That was a big red flag to me years ago when we started working on this project."
SEE ALSO: Study: Most Newborns with Epilepsy Benefit from Genetic Testing
To provide further evidence that SCN1A mutation was wreaking havoc in the heart, Parent and Isom's team used CRISPR-Cas9 technology to molecularly delete the gene from the cells of a healthy child without epilepsy. When the researchers repeated their experiment with those cells, they saw the same increase in sodium current.
Next, Isom and Parent plan to look at different genetic mutations related to SUDEP and at the potential use of repurposed drugs to treat Dravet syndrome and other forms of epilepsy.
"This is personalized medicine," Isom says. "This is what we're all after in the grand scheme of things. It takes a long time and a lot of money, but it works. If we can help one child, then it's worth it."
The study was co-authored by Chad Frasier, Ph.D., of the Department of Pharmacology, and Helen Zhang of the Department of Neurology. The work is supported by National Institutes of Health grants T32HL007853, UL1TR000433, R37-NS076752 and U01-NS090364.

Explore a variety of healthcare news & stories by visiting the Health Lab home page for more articles.

Department of Communication at Michigan Medicine
Want top health & research news weekly? Sign up for Health Lab’s newsletters today!